Short Answer: Identification reliability varies enormously depending on the species, the reference database, and the sequencing approach. For well-studied vertebrates with complete reference databases, airborne eDNA identifications are generally reliable at genus level and more often also at species level. For insects, rare plants, and taxa in under-sampled regions, the reference database is the binding constraint, and identifications must be treated with more caution. The biggest source of misidentification is not the eDNA method itself, but the incompleteness of the databases it relies on.
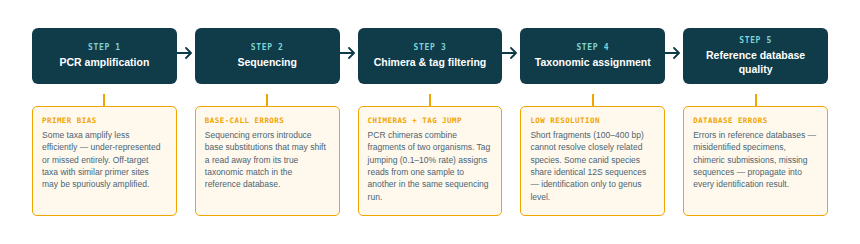
1. The identification pipeline and where errors enter
This article is specifically about what can go wrong in the analytical process once DNA has already entered the sample: not about whether the right species were present, nor about field factors such as transport distance or temporal persistence that affect which organisms' DNA reaches a sampler. The question here is: given that a DNA molecule from a species is on the filter, how reliably can the laboratory and bioinformatics pipeline correctly assign it to that species?
The answer depends on a sequential pipeline, and errors can be introduced at any stage:
-
1DNA amplification(PCR): primer bias can cause some taxa to amplify more efficiently than others
-
2Sequencing:sequencing errors introduce base substitutions that may cause misassignment
-
3Bioinformatic filtering:chimeric sequences (artefacts from PCR that combine fragments of two different organisms) must be detected and removed
-
4Taxonomic assignment:the sequenced fragment is matched against a reference database; if the correct species is absent or its sequence is similar to another species, misidentification occurs
-
5Database quality:errors or misidentifications in the reference database propagate into eDNA results
In much of scientific literature the reference database incompleteness and quality is mentioned as the dominant source of misidentification in current practice.
2. The reference database constraint
2.1 Coverage is highly uneven
Sullivan et al. (2025) found that a large majority of sequencing reads from their Swedish boreal ecosystem could not be classified to any known taxon. As databases improve, the same samples or their derived and stored digital sequences can be re-analysed to reveal what these reads represent.
Database coverage is highest for:
- Vertebrates in Europe and North America (where most reference sequencing has been done)
- Economically important species (crop plants, protected and invasive species, pets, game animals, disease vectors)
- Well-studied fungal genera in temperate regions
Database coverage is lowest for:
- Invertebrates, particularly in tropical regions
- Plants outside Europe and North America
- Bacteria and archaea at species level
- Any taxon in under-sampled regions
2.2 Database errors propagate
Reference databases themselves contain errors: sequences deposited under incorrect species names, chimeric sequences submitted inadvertently, and sequences from misidentified museum specimens. These errors are impossible to detect without extensive verification. Tournayre et al. (2025) explicitly acknowledged that some of their detections, including potentially invasive fish species, "should be interpreted with caution as we anticipate errors in reference collections."
The quality of eDNA identifications is fundamentally limited by the quality of the databases used. High-quality curated databases such as BOLD (partially curated) and species-specific national barcode libraries, or databases built for regulatory monitoring, produce more reliable results than general-purpose databases like NCBI GenBank, which accepts sequences from any depositor with minimal quality control.
3. Primer-related misidentification
Metabarcoding uses universal primers designed to amplify a target gene from all members of a broad taxonomic group. In practice, no primer pair amplifies all taxa in a group with equal efficiency. Taxa with primer binding site mismatches are amplified less efficiently (or not at all), which can cause them to be under-represented or missed entirely.
Conversely, non-target taxa that have sequences similar to the primer binding site may be spuriously amplified. These off-target amplifications can produce reads that match real species in the database but represent artefactual detections.
Careful primer validation against a diverse reference panel before deployment can reduce but not eliminate this problem. The shift towards shotgun metagenomics (which does not use target-specific primers) avoids primer bias entirely, at the cost of higher sequencing depth and computational resources.
4. Short barcode fragments and species resolution
Most airborne eDNA metabarcoding uses very short DNA fragments: typically 100–400 base pairs for metabarcoding, compared to the 600+ base pairs of the full standard barcode for animals (COI). Short fragments provide less taxonomic resolution.
For vertebrates using the 12S rRNA mini-barcode (commonly 100–170 bp), species-level resolution is achievable for most mammals and birds in regions with good reference coverage. For some groups, particularly certain canid species (dogs, foxes, wolves) that share very similar 12S sequences, species-level resolution is not possible from short fragments, and identification is reported at genus level (Canis sp.).
For plants using ITS1/ITS2 barcodes, species-level resolution varies considerably by taxonomic group. Some genera are well-resolved; others have ITS sequences so conserved that congeners are indistinguishable. This is well-documented in the palynology literature for pollen identification (Bell et al. 2016; San Martin et al. 2024).
5. Chimeras, PCR artefacts & tag jumping
During PCR amplification, incomplete extension products from one cycle can prime off a different template in the next cycle, producing hybrid sequences (“chimeras”) that do not match any real organism. Chimera-checking algorithms (implemented in tools like USEARCH, UCHIME, and DADA2) detect and remove most chimeras during bioinformatic processing, but this step introduces its own uncertainty: genuine low-abundance sequences can occasionally be misclassified as chimeric and discarded.
Deep mixtures of many taxa in a single sample, as in airborne eDNA from diverse communities, increase chimera formation rates compared to simpler samples. Careful optimisation of PCR conditions (reduced cycle number, high-fidelity polymerase, appropriate annealing temperature) minimises but does not eliminate chimera production.
Tag jumping (also called index switching or index bleed) is a sequencing-stage artefact that deserves specific attention in airborne eDNA work. When multiple samples are multiplexed on a single sequencing run, as is standard practice for cost efficiency, each sample is identified by a unique combination of short DNA tags (indices) ligated to the library molecules. Tag jumping occurs when these indices swap between molecules during the library preparation or sequencing process, causing a read from one sample to be misassigned to another.
The consequence in airborne eDNA is a false positive: a species genuinely present in a high-concentration sample (such as a zoo animal or abundant agricultural species) can appear as a phantom detection in an adjacent low-concentration environmental sample that shares the same sequencing lane. In multiplex runs with many samples, typical for large monitoring programmes, even a jumping rate of 0.1–1% of reads can be sufficient to generate plausible-looking but spurious detections.
Rodriguez-Martinez et al. (2025) specifically investigated tag jumping in a metabarcoding context and confirmed that T4 DNA polymerase, commonly used in library preparation end-repair steps, is a major contributor to the process, because it can extend overhanging ends and thereby facilitate index transfer between molecules. They showed that combinatorial tagging protocols (where each sample receives a unique combination of two different indices, one on each strand) are substantially more susceptible to jumping than twin-tagging protocols (where both strands of a molecule receive identical indices), because a jumped molecule under twin-tagging is identifiable as a contaminant by its non-matching index pair.
Mitigation strategies include: (1) using dual-index (combinatorial) libraries with a sufficient diversity of index combinations that spurious assignments remain statistically distinguishable; (2) applying a minimum read-count threshold per taxon per sample: taxa appearing with very few reads in a sample are more likely to be tag-jump artefacts than genuine detections; (3) sequencing negative controls (blank libraries prepared with no template DNA) per batch to estimate the background jump rate; and (4) using unique molecular identifiers (UMIs) which tag individual molecules before amplification, allowing true duplicates to be distinguished from jumped reads after sequencing.
6. When identifications are most and least reliable
Most reliable conditions:
- Species has multiple high-quality reference sequences in the database
- Species is genetically distinct from close relatives at the target barcode locus
- Sequencing depth is high (many reads provide statistical confidence)
- Multiple samples or replicates show consistent detection
- Metabarcoding primer pairs have been validated for the target group
Least reliable conditions:
- Species has no reference sequence, or only one low-quality sequence
- Species is closely related to others with similar barcode sequences
- Sequencing depth is low (few reads, high uncertainty)
- Single detection in a single sample
- Target region has known amplification bias for the taxon group
Strictly speaking, a species without any reference sequence cannot be identified at all: its DNA may be present in the sample and will produce sequencing reads, but those reads will not match anything in the database and will be discarded or left unclassified. Usually most algorithm will classify to a higher taxonomic level is (like genus or family) if a particular certainty level in the match with a reference sequence can not be achieved.
7. Practical guidance for interpreting identifications
- For well-databased vertebrates (birds, mammals in Europe):Species-level identifications are generally reliable. Corroborate unexpected detections with other evidence.
- For insects and invertebrates:Treat identifications with considerable caution unless validated against a curated reference database. Genus-level identification is more reliable than species level.
- For plants:Identifications to genus are generally reliable; species-level resolution depends strongly on the taxonomic group and whether the specific region’s flora is well-represented in the reference database.
- For potential invasive species:Apply heightened scrutiny. The consequences of acting on a false positive (unnecessary management intervention) or false negative (missed invasion) are both significant. Require corroborating evidence before management action.
- For any single-detection anomaly:Treat as a hypothesis to test, not a confirmed observation. Replicated detections across multiple sampling events are far more credible.
8. The improving trajectory
The reference database limitation is not permanent. Several major initiatives are expanding coverage:
This means identifications from airborne eDNA will become more reliable over time without any change to the original samples.
References
- Alberdi A et al. (2018). Scrutinizing key steps for reliable metabarcoding. Methods in Ecology and Evolution 9:134–147. https://doi.org/10.1111/2041-210x.12849
- Bálint M et al. (2018). Environmental DNA time series in ecology. Trends in Ecology & Evolution 33:945–957. https://doi.org/10.1016/j.tree.2018.09.003
- Bell KL et al. (2016). Pollen DNA barcoding: current applications and future prospects. Genome 59:629–640. https://doi.org/10.1139/gen-2015-0200
- Berelson MFG et al. (2025). From air to insight: the evolution of airborne DNA sequencing technologies. Microbiology 171:001564. https://doi.org/10.1099/mic.0.001564
- Goldberg CS et al. (2016). Critical considerations for the application of environmental DNA methods to detect aquatic species. Methods in Ecology and Evolution 7:1299–1307. https://doi.org/10.1111/2041-210X.12595
- Rodriguez-Martinez S et al. (2025). Tag jumping produces major distortion on metabarcoding-based reconstructions of past and present environments. Environmental DNA 7:e70148. https://doi.org/10.1002/edn3.70148
- San Martin G et al. (2024). How reliable is metabarcoding for pollen identification? PeerJ 12:e16567. https://doi.org/10.7717/peerj.16567
- Sullivan AR et al. (2025). Airborne eDNA captures three decades of ecosystem biodiversity. Nature Communications 16:11281. https://doi.org/10.1038/s41467-025-67676-7
- Tournayre O et al. (2025). First national survey of terrestrial biodiversity using airborne eDNA. Scientific Reports. https://doi.org/10.1038/s41598-025-03650-z